Posted on June 21, 2022
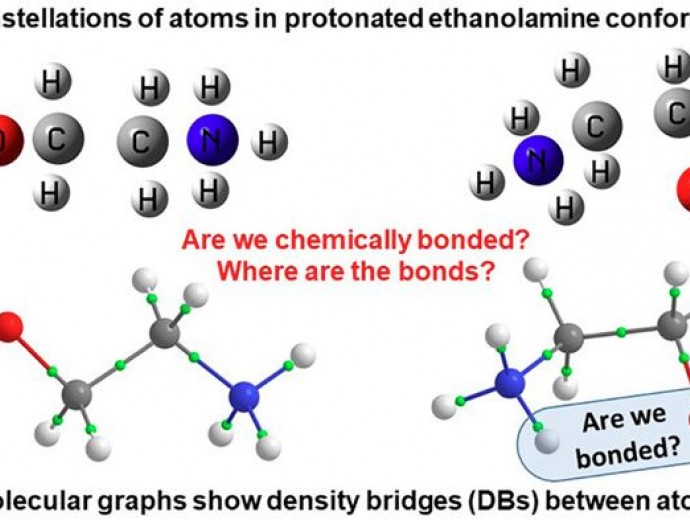
Chemical bonding is at heart but, not being a quantum mechanical-defined physical property of a system, is a subject of endless and often fruitless debates. Having so many and very different models of chemical bonding without knowing what this really is does not make it easier. There is, however, a general agreement that concentrating electron density (ED) in and delocalizing ED to internuclear region is always associated with minimizing system's energy and synonymous with chemical bonding. Fragment, atomic, localized, delocalized, and interatomic (FALDI)-based density analysis involves entire space occupied by a molecule. From this molecular-wide and density-based methodology, it is possible to quantify localized and delocalized by all atoms ED at any coordinate r, including critical points on Bader's molecular graphs. Each atom and atom-pair contributions of delocalized density are quantified to reveal major players in the all-atom and molecular-wide chemical bonding. Partitioning the total ED to individual molecular or natural orbital's contributions using MO-ED and MO-DI methods, in conjunction with one dimensional (1D) cross section methodology, generates an orbital-based molecular-wide picture. This provides, besides reproducing results from FALDI, qualitative description of orbitals' nature that correlates well with classical understanding of bonding, nonbonding, and antibonding orbitals. A qualitative and quantitative impact of an immediate, distant, or molecular-wide molecular environment on intra- and intermolecular di-atomic, intra- and interfragment interactions is the domain of the Fragment Attributed Molecular System Energy Change (FAMSEC) family of methods. The FALDI, FAMSEC, MO-ED, MO-DI, and 1D cross section methodologies provide consistent and quantifiable physics-based picture of molecular-wide chemical bonding without invoking unicorns, such as a chemical bond.
Read more: https://doi.org/10.1002/wcms.1579




Copyright © University of Pretoria 2024. All rights reserved.




 Virtual Campus
Virtual Campus
Get Social With Us
Download the UP Mobile App